












应用领域
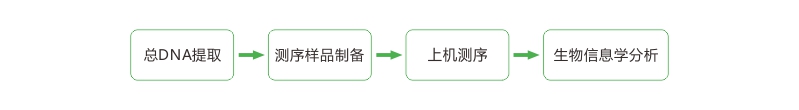
技术路线
分析内容
|
1.多态SNP标记筛选
1.1亲本间多态标记筛选
1.2子代基因分型和标记过滤
1.3有效遗传标记筛选
2.遗传图谱构建
2.1上图标记完整性分布统计
2.2连锁群构建
2.3连锁图绘制
|
3.图谱质量评估
4.QTL分析
4.1 基于CIM模型的QTL分析
4.2显著位点的筛选
4.3基于QTL分析结果绘图
4.4基于2-LOD置信区间的QTL区间定位
4.5 QTL区间内相关候选基因的功能注释;(必须有参考基因组)
|
多遗传图谱整合组装高质量梨树基因组
研究材料
研究内容
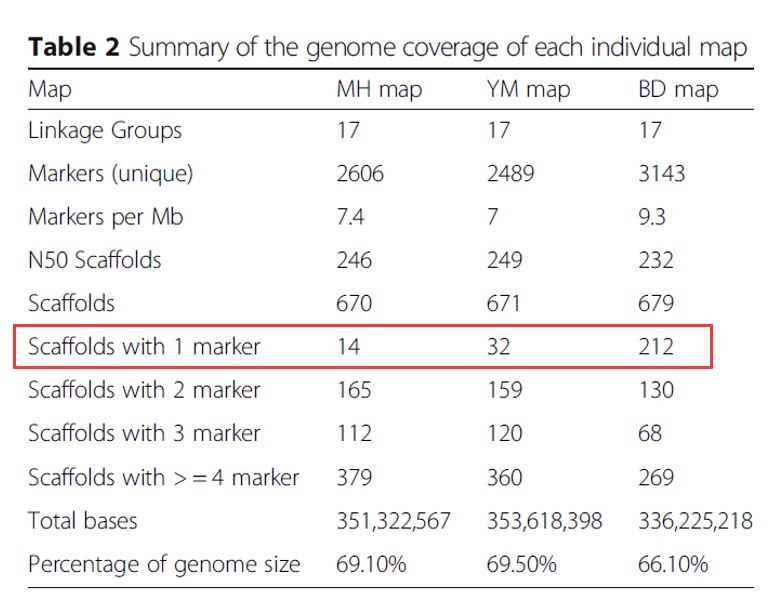
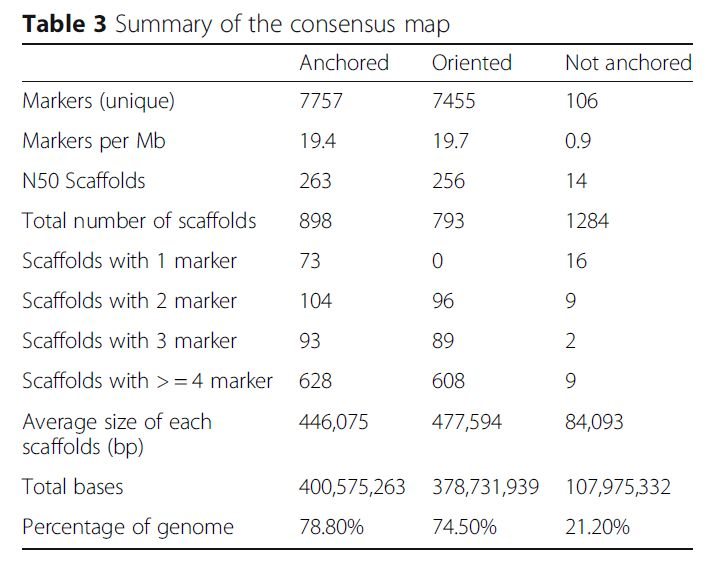
各个图谱的概况
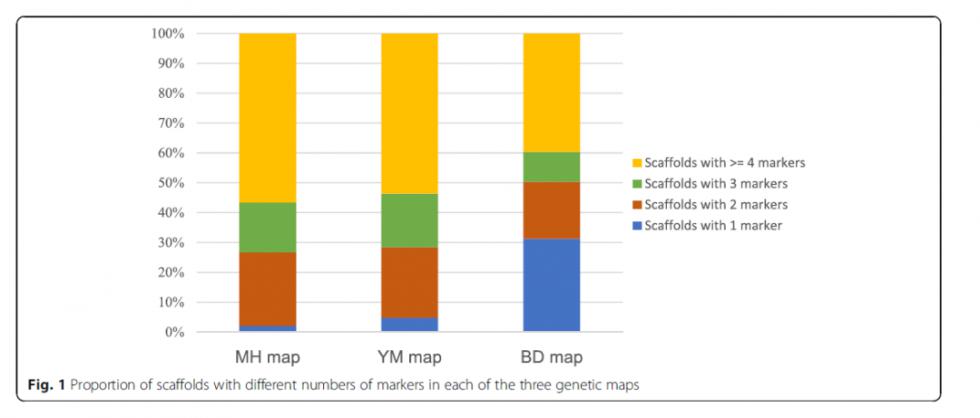
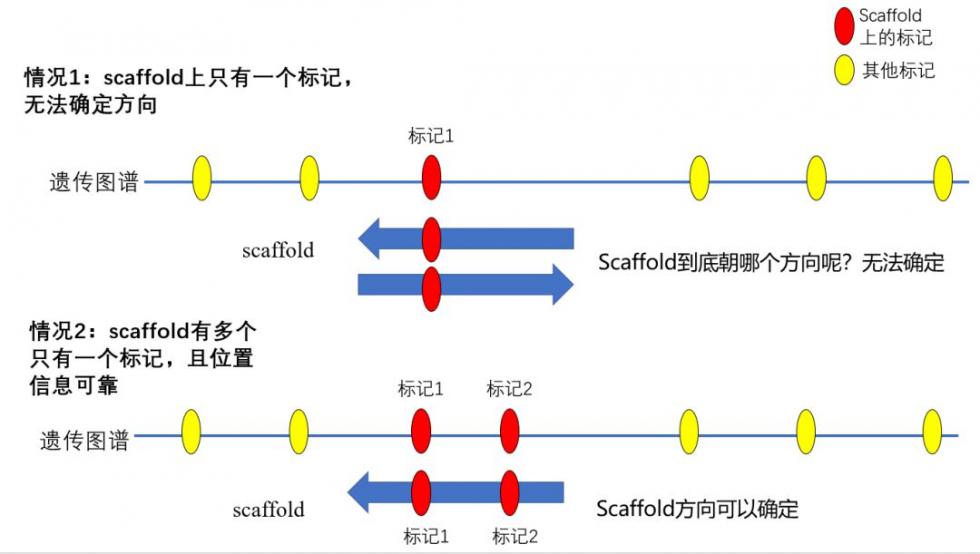
图谱整合
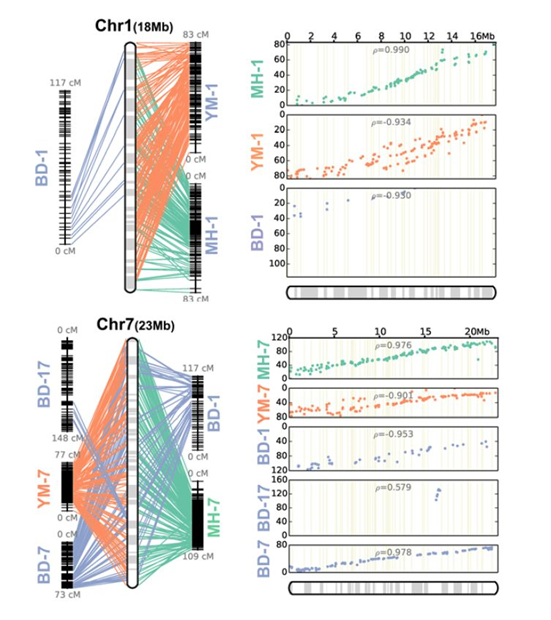
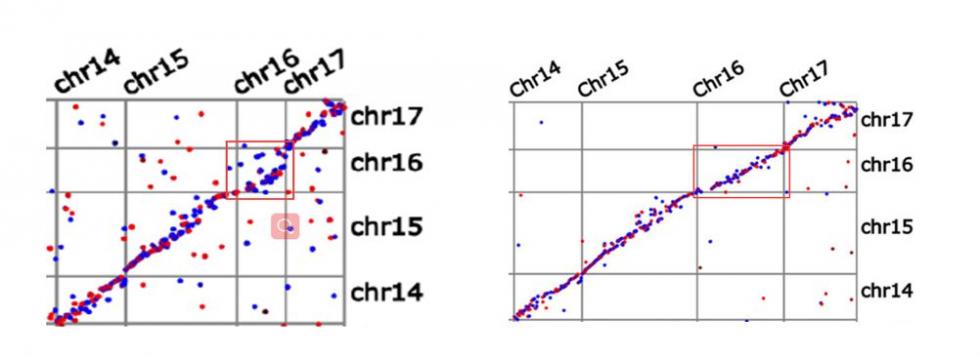
基于整合图谱的3个子图谱比较


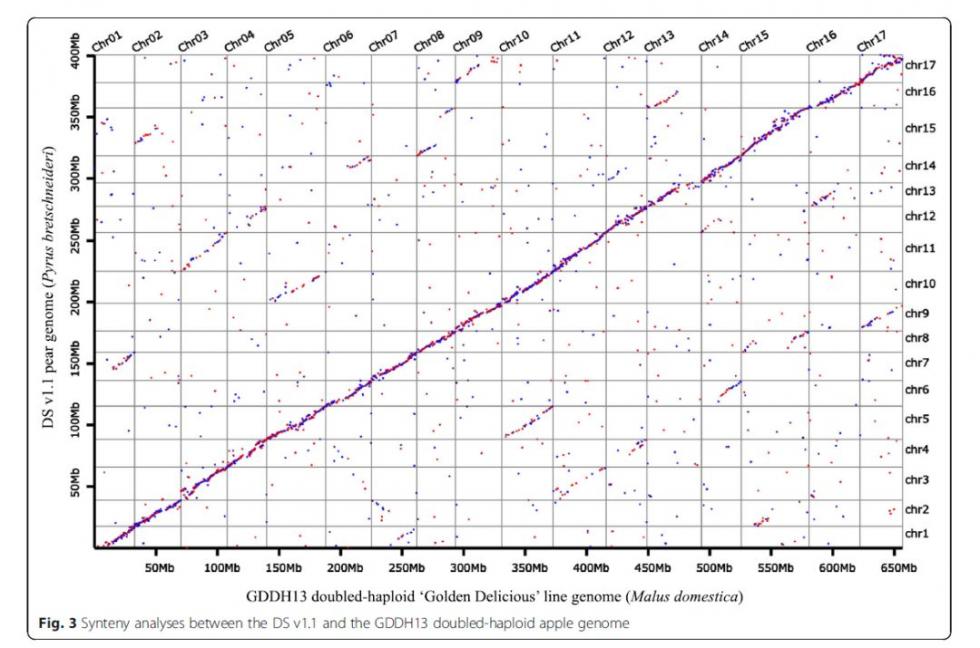
与苹果的共线性比较
小结


