












应用领域
技术路线

分析内容
|
标准信息分析
1. 测序质量评估与数据过滤
2. 过滤宿主数据(若有宿主)
3. 宏基因组组装
4. 组装结果统计与评估
5. 物种注释及物种丰度统计
物种分布堆叠图
物种分布circos图
物种分布热图
6. 基因预测
7. 基因丰度分析
8. Core-pan曲线分析
9. 基因数据库功能注释
KEGG、eggNOG、CAZy、PHI、CARD、VFDB
10. 基因功能丰度热图、柱形图、circos图
11. 多样品间比较分析(基于各物种分类、功能分类)
a) 组间比较韦恩图
b) 组间比较upset图
c) 样本关系热图
d) UPGMA聚类树分析
|
e) PCA分析
f) PCoA分析
g) NMDS分析
h) Adonis检验
i) Anosim检验
12.差异分析(基于各物种分类、功能分类)
a) Welch’s T-test组间差异分析
b) Anova组间差异分析
c) 物种富集三元图
e) Metastates分析
f) Lefse分析
定制化信息分析
1. binning分析
2. CAG分析
3. 肠型分析
4. SEM结构方程分析
|
宏基因组测序探讨自闭症与肠道微生物的关联
研究背景
实验方法
研究思路
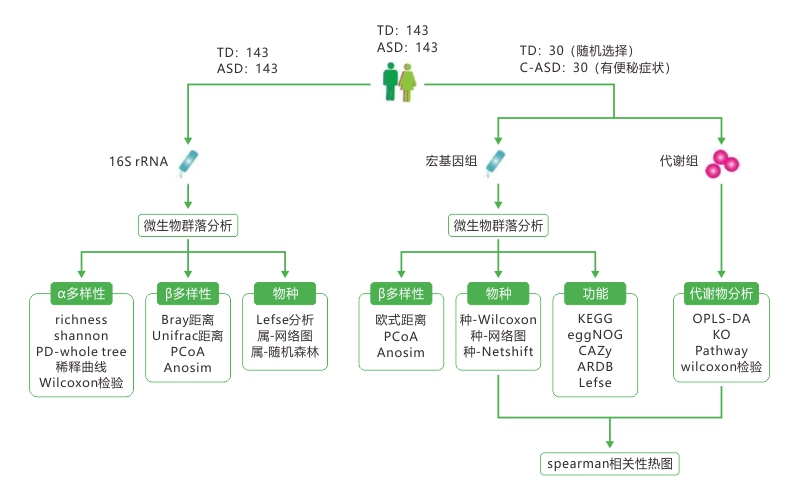
研究结果



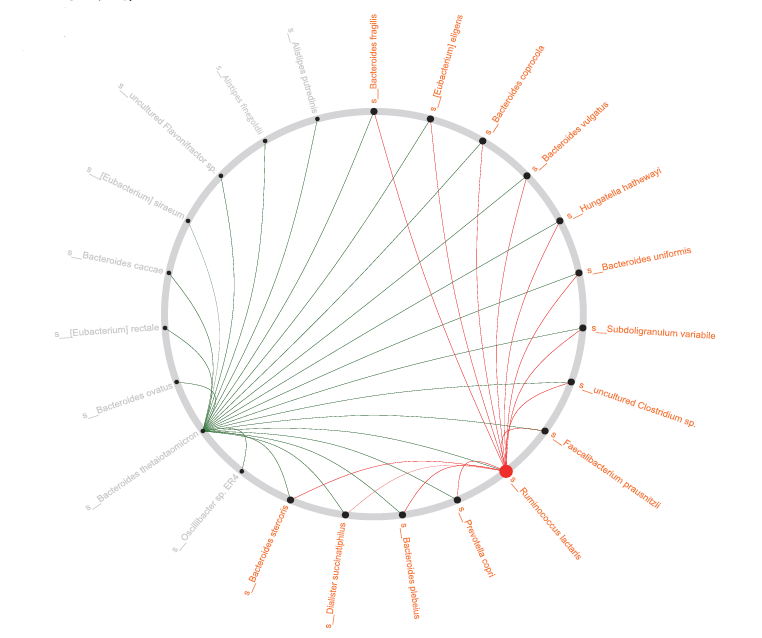

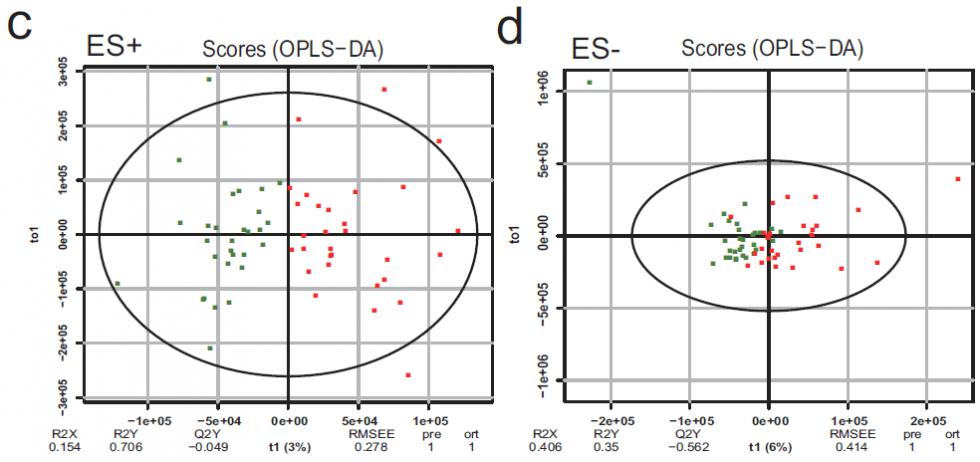
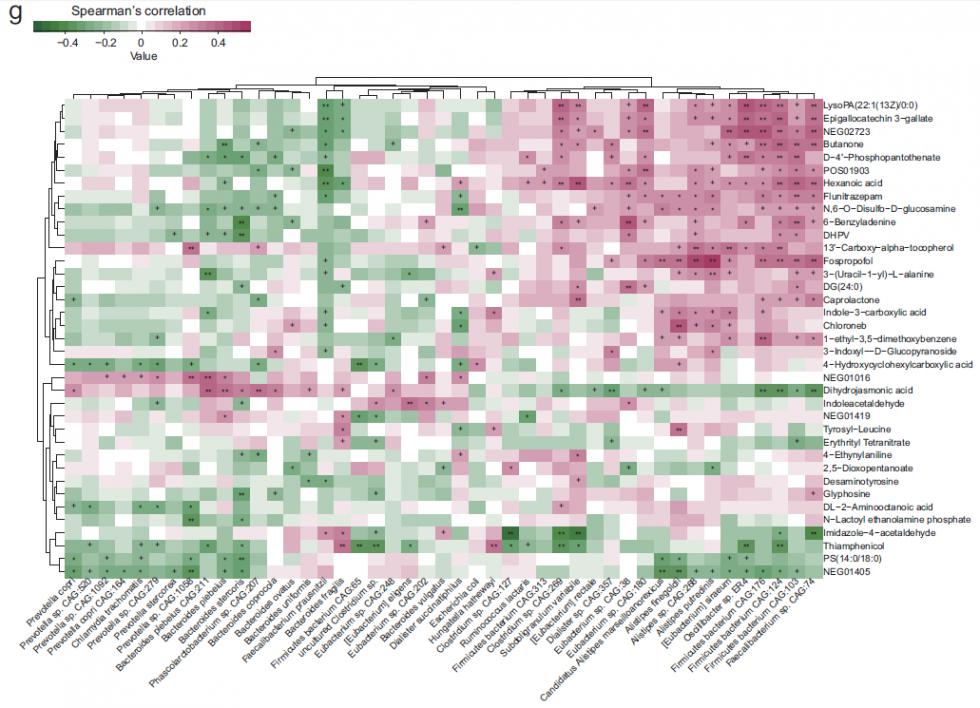
小结
参考文献


